
![]()
![]()
Table des Matières :
L'ensemble de l'ADN d'une cellule est appelé génome. Une vidéo (en anglais: 'What is genomics?') pour mieux comprendre la notion de 'génome'.
En 2001, les revues Nature et Science publiaient respectivement une première analyse de la séquence 'complète' de 2 génomes humains (23 chromosomes; longueur totale: ~3'000'000'000 bp).
1. Le génome de référence (par le consortium publique)
2. Le génome de Craig Venter (par un consortium privé, Celera)
Où trouver les séquences?
Les séquences de ces 2 génomes ont été soumises aux banques de données publiques ( EMBL/GenBank/DDBJ : http://www.insdc.org/ ).
- Le génome humain de référence Ce génome est 'haploide': une séquence est disponible (mélange des chromosomes paternels et maternels de ~10 individus anonymes); un seul brin (forward strand);
* Séquence du chromosome X humain @ GenBank
* Séquence du génome de la mitochondrie (Homo sapiens; reference) @ GenBank
* Séquence (partielle) du chromosome 1 humain @ GenBank
- Le génome 'Celera' : 6 individus dont Craig Venter "DNA Donor Name: J. Craig Venter | Date of Birth: October 14, 1946 | Sex: Male | Ethnicity: Caucasian | Descent: European - England" Ce génome est 'diploide': la séquence des allèles respectivement paternel et maternel est connue; une seule séquence est disponible: les différences sont répertoriées.
* Séquence du chromosome X humain (Celera) @ GenBank
Info: ~3,213,401 polymorphismes (différences avec le génome complet de référence) ont été identifiés ( Levy et al., 2007 )
Ce site propose une bonne vue d'ensemble des séquences de génomes humains disponibles: Genomes @ NCBI (section genome)
Autres génomes humains
- Nouveaux projets:
séquencer différentes cellules d'un même individu (lymphocyte B, T, cellules sexuelles, melanocytes...). But: avoir une liste des mutations héritées et des mutations somatiques
NB: Toutes les différences entre les séquences des génomes disponibles sont rapportées à la séquence du génome humain de référence.
Au niveau moléculaire, ces différences s’appellent des SNPs ( SNP: single nucleotide polymorphism ). Le terme « polymorphisme » désigne en général un locus avec plusieurs allèles ; dans ce cas, il désigne un site de l’ADN avec plusieurs nucléotides (ou site variable).
Il existe environ 10 millions de SNPs chez les humains; un nucléotide sur 1 200, en moyenne, varie d’une personne à l’autre (source: hapmap ). On parle plus spécifiquement de polymorphisme lorsqu’un SNP est présent dans au moins 1% de la population.
On appelle mutation le processus moléculaire produisant un changement ponctuel de nucléotide au niveau de la séquence d’ADN. Les mutations génèrent donc des SNPs.
Les mutations peuvent se produire dans des régions codantes (transcrites en ARN messager, et généralement traduites en protéines) ou non codantes . Elles peuvent donc induire, ou non, un changement au niveau de la séquence en acides aminés d’une protéine.
Les polymorphismes peuvent donc se situer dans des régions de l’ADN fonctionnellement importantes car elles codent pour des protéines essentielles à la survie ou au bon fonctionnement de l’organisme. Certains polymorphismes sont alors létaux ou associés à des maladies .
Mais la grande majorité des polymorphismes sont neutres : ils n’affectent pas l’individu. Une immense quantité de variation génétique neutre de notre ADN existe donc dans toutes les populations.

Les polymorphismes ou variants peuvent être dans des régions codantes ou non codantes (pour des protéines). Les polymorphismes peuvent être neutres ou induire un changement au niveau de la séquence en acides aminés par exemple.
On parle parfois de mutation , lorsque le polymorphisme est associé avec une maladie. Une mutation est en fait le processus qui génère les variants.
Nous ne parlerons pas dans ce TP des différences impliquant des délétions, insertions, duplications etc. qui sont notamment répertoriées dans dbVar
Projet 1000genomes: http://www.1000genomes.org/
'A Deep Catalog of Human Genetic Variation'
'The goal of the 1000 Genomes Project is to find most genetic variants that have frequencies of at least 1% in the populations studied. This goal can be attained by sequencing many individuals lightly. To sequence a person's genome, many copies of the DNA are broken into short pieces and each piece is sequenced. The many copies of DNA mean that the DNA pieces are more-or-less randomly distributed across the genome. The pieces are then aligned to the reference sequence and joined together. To find the complete genomic sequence of one person with current sequencing platforms requires sequencing that person's DNA the equivalent of about 28 times (called 28X). If the amount of sequence done is only an average of once across the genome (1X), then much of the sequence will be missed, because some genomic locations will be covered by several pieces while others will have none. The deeper the sequencing coverage, the more of the genome will be covered at least once. Also, people are diploid; the deeper the sequencing coverage, the more likely that both chromosomes at a location will be included. In addition, deeper coverage is particularly useful for detecting structural variants, and allows sequencing errors to be corrected.'
3 études pilotes sont en cours:
Vous aurez accès aux données des 2 trios CEU et YRI.
Projet HapMap: www.hapmap.org/
'Le projet international HapMap est une initiative visant à relever et à cataloguer les similitudes et les différences génétiques entre les humains.... pour permettre aux chercheurs de découvrir les gènes qui jouent un rôle dans la santé, la maladie et la réponse des individus aux médicaments et aux facteurs environnementaux.' plus d'infos...
Projet ENCODE
ENCyclopedia Of DNA Elements: identifier les éléments fonctionels du génome humain. Une première étape à consister à reanalyser une partie du génome humain (1%) (séquence, expression des gènes, etc.).
La banque de données dpSNP répertorie les polymorphismes humains. Les polymorphismes liés aux maladies sont rares dans les populations humaines (ouf !)....On ne trouve donc pas facilement des infos sur leur fréquence. dbSNP contient donc surtout des polymorphismes 'neutres', mais très peu de 'mutations'.
Chaque entrée (polymorphisme) de dbSNP a un numéro d'accession du type 'rs12913832'.
Exemple: rs12913832 un allèle HERC2 fortement impliqué dans la coloration bleue des yeux: Allèle ancestral: a Mutation a -> g Génotype possible: aa, ag, gg L'allèle g est plus fréquent dans les populations avec des yeux bleus (nord de l'Europe par exemple)
Ce polymorphisme se trouve proche du gène HERC2, l’un des "gènes" impliqués dans la pigmentation des yeux.
Attention au sens dans lequel le gène est lu par rapport à la séquence du génome de référence (un polymorphiosme T->G sur le brin forward peut être vu comme A-> C sur le brin reverse )!!!
OMIM répertorie tous les gènes et maladies humaines, inclues les mutations.
Exemple:
HERC2
, le 'gène' des yeux bleus.
On accède à cette banque de données depuis dbSNP (le lien est tout en bas de la page, mais il n'existe pas pour tous les polymorphismes). Chercher le mot 'Alfred' dans la page grâce à 'Ctrl/Cmd F'
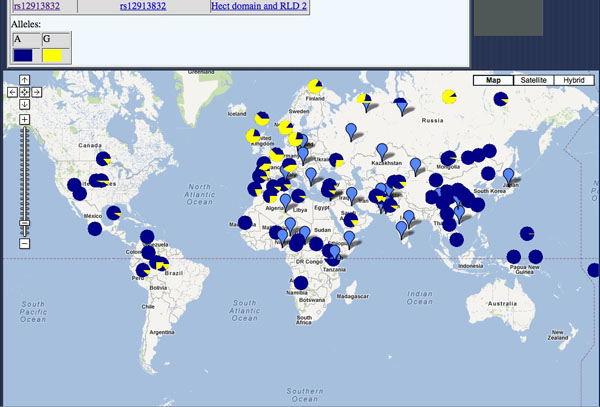
Exemple: rs12913832 , l'allèle des yeux bleus.
On accède aussi directement depuis la banque de données Alfred (copier coller le no dbSNP, par exemple rs12913832)
Exemple: Alfred -> ce SNP rs12913832 Puis cliquer sur google map...
Les 'genome browser' permettent de se balader dans les génomes et d'accéder à une montagne d'information, inclues les variations génétiques (un peu comme Google map).
Rappel: Toutes les différences entre les séquences des génomes disponibles sont rapportées à une séquence de référence du génome humain .
Il existe plusieurs 'Genome browser'
Nous vous proposons de découvrir celui de UCSC
Copier coller le nom du gène HERC2 dans le champ 'gene' puis cliquer le bouton submit
![]()
Il y a beaucoup d'information disponible: c'est utile de choisir celles qui nous intéressent, comme par exemple:
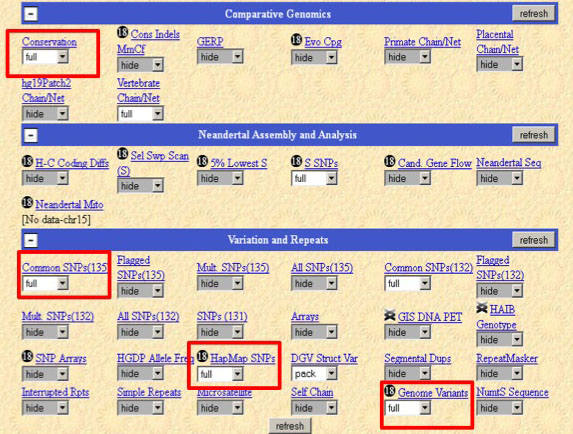
Cliquer sur refresh (une fois sélectionnées, les sections restent visibles par défaut)
Vous voyez apparaître tous les polymorphismes connus pour ce gène (et présents dans plus de 1% de la population), inclus ceux de Craig Venter, James Watson et des 1000 génomes projects
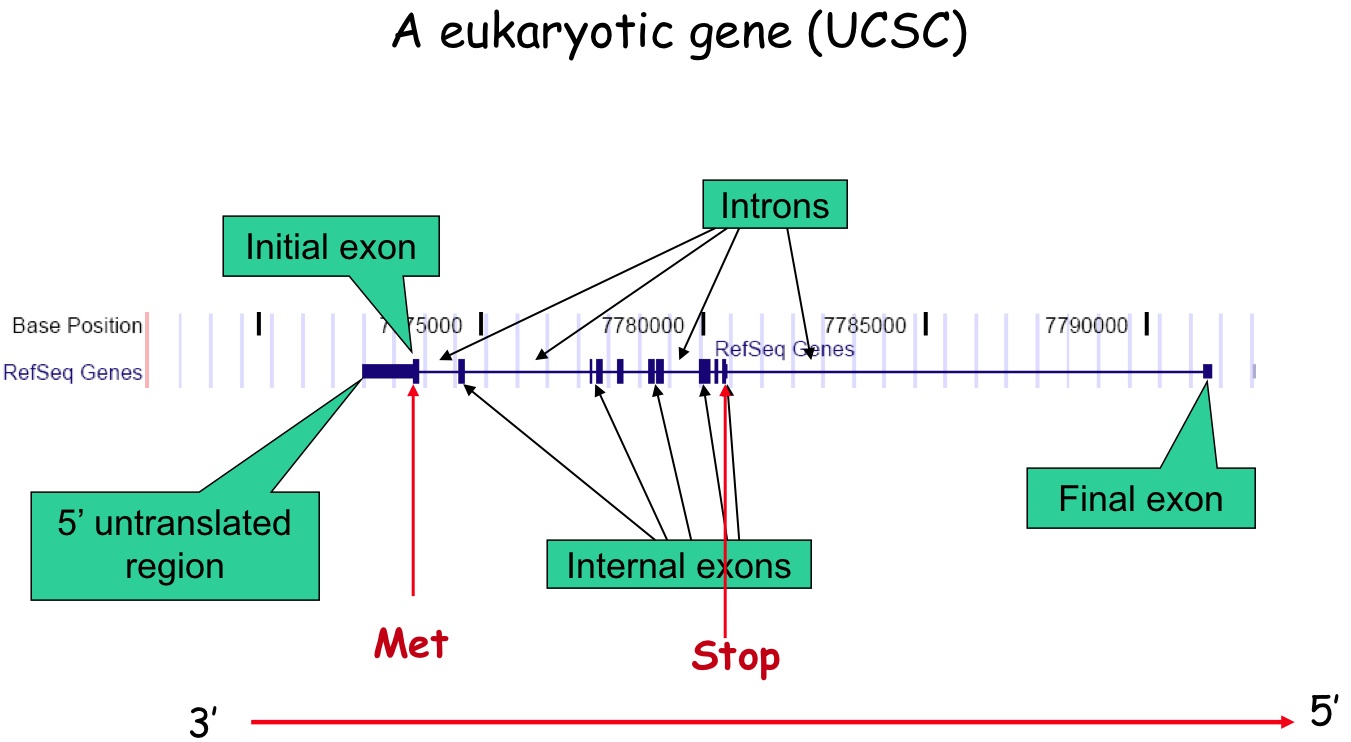
On peut mettre en évidence que les polymorphismes sont répartis entre les régions codantes (exons) et non-codantes (introns)

Sur la ligne du haut on voit les positions sur le Chr 15. Sur la deuxième ligne, la structure du gène :les barres épaisses sont les exons ( ici très éparpillés) et les < ou > déterminent le sens de lecture. La troisième ligne indique la répartition des SNP.
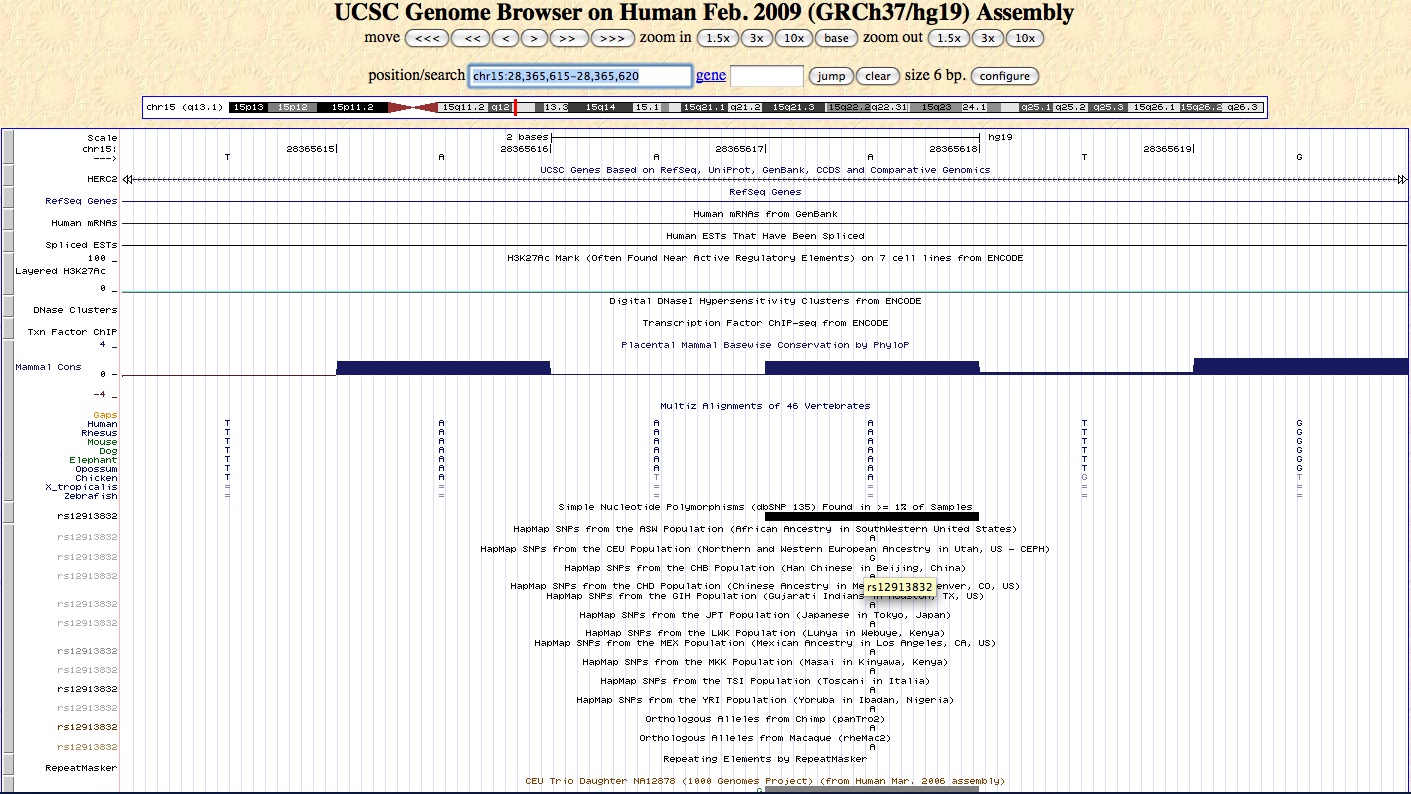
Un SNP fortement impliqué dans la coloration bleue des yeux est situé à la position 15: 28365618 (sur le génome de référence)
introduire ce chiffre dans le champ 'position' en haut de la page : chr15:28365618
Remarque: vous pouvez aussi chercher directement avec le no 'rs12913832'; l'output ne sera toutefois pas tout-à-fait pareil.
Vous voyez apparaître tous les polymorphismes connus pour ce gène (et présents dans plus de 1% de la population), inclus, par exemple, ceux de Craig Venter et des 1000 génomes projects.
Faire un zoom out 10x pour voir apparaître la séquence de l'ADN dans son voisinage. (Ou indiquer chr15:28,365,615-28,365,620)
Dans la base de données ALFRED coller le numéro de SNP rs12913832
Solution ici, mais il faudra cliquer sur Google maps
Notez bien que l'allèle avec un G est associé au phénotype Bleu ... mais c'est l'allèle A qui est représenté sur la carte en bleu !
Très malencontreux.
Alicia Sanchez-Mazas rappelle que :
"A l'examen de ce tablaau on pourrait avoir l’impression que tout est qualitatif, déterminé par des mutations ponctuelles.
Or la plupart des caractères phénotypiques sont quantitatifs, déterminés par de nombreux gènes qui interviennent dans des régulations d’expression. On a aussi l’impression que tout est déterministe. Or l’environnement agit aussi de manière complexe dans l’expression. L’une des grandes voies de recherche actuelles est précisément d’identifier toutes ces régulations au niveau cellulaire. Cela nous éloigne beaucoup de la simple génétique de Mendel !"
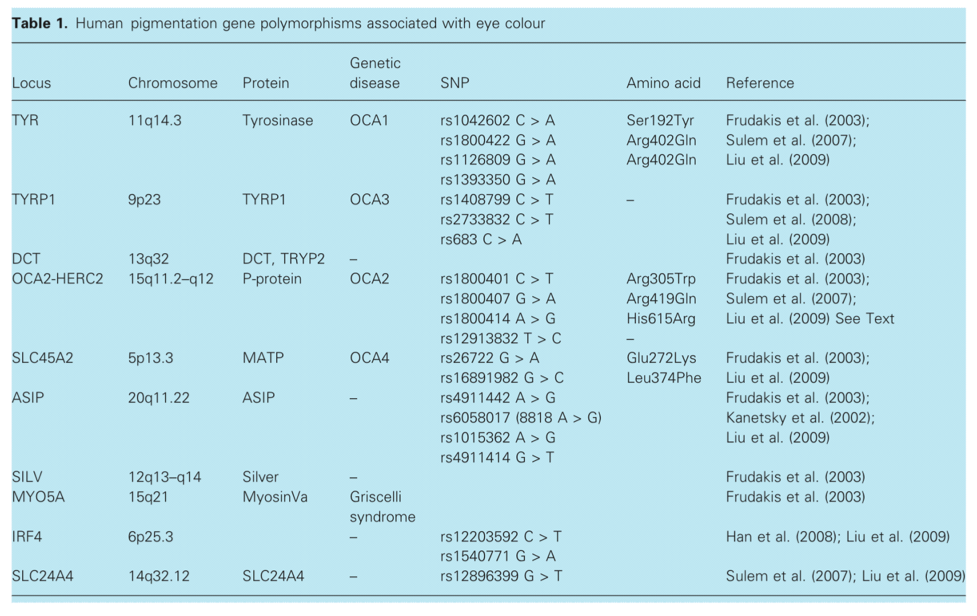
| Gene | db SNP AC | polymorphism (DNA) | polymorphism (protein) | N° chromosome et position * | Fonction | UniProtKB AC | Alfred |
| HERC2 | rs12913832 | a->g | non codant | 15: 28365618 | yeux bruns- > bleus (PubMed) | O95714 | Alfred (allèle yeux bleux plus fréquent au nord de l'Europe) |
| PAH | pas de polymorphism neutre; pas de lien vers dbSNP | plusieurs sites | Plusieurs sites: annotations des variants dans UniProtKB/Swiss-Prot | chr12:103232104-103311381 | Phenylketonuria (PKU) OMIM | P00439 | pas disponible |
| MRP8 | rs17822931 | c->t sur le brin reverse
g->a sur fwd |
G->R | 16: 48258198 | cire d'oreille humide -> sèche | Q96J66 | Alfred (allèle cire humide plus fréquent en Asie) |
| CYP2C9 | rs28371686 | c->g | D->E | 10: 96741058 | 'metaboliseur' lent -> rapide | P11712 | Alfred |
| CYP2C9 | rs1799853 rs1057910 rs9923231 |
10 | 'metaboliseur' lent -> rapide (testé pour l'anti-coagulant warfarin) | P11712 | Alfred (rs1799853) | ||
| HBB | rs334 | a->c a->t |
E->A E->V |
11: 5248232 | sickle cell anemia (anémie falciforme) | P68871 | non disponible (maladie); plus fréquent en Afrique et en Arabie Saoudite |
| CFTR | rs35516286 | t->c | I->T | 7: 117171122 | Mucoviscidose | P13569 | non disponible (maladie) |
| KL (Klotho) (1) | rs9536314 | t->g | F->V | 13: 33628138 | Homozygosity for KL-VS allele is associated with decreased longevity and increased cardiovascular disease risk. | Q9UEF7 | Alfred (?) |
| KL (Klotho) (2) | rs9527025 | t->g | C->S | 13: 33628193 | Homozygosity for KL-VS allele is associated with decreased longevity and increased cardiovascular disease risk. | Q9UEF7 | non disponible |
| p53 | rs1042522 | g->c | P->R | 17: 7579472 | frequent polymorphism in colonic cancer patients | P04637 | non disponible (maladie) |
| SLC24A5 | rs1426654 | a->g | T->A | 15: 48426484 | coloration de la peau (variation continue*2) | Q71RS6 | non disponible |
| DARC | rs12075 | a -> g | non codant (mutation in the promoter) | 1: 159175354 | malaria resistance (g) | Q16570 | non disponible |
| APOE | rs429358 ApoE4 allele = rs429358(C) + rs7412(C) can raise the risk of Alzheimer's disease by more than 10x |
(t -> c) + (t -> c) | ApoE*4: 112: C -> R + 172: R -> C | 19: 45411941 + 19: 45412079 | Transport des lipides; E*4 -> Alzheimer précoce (MIM 'Craig Venter' est respectivement C/T et C/C ... |
)
UniProtKB P02649 | Alfred |
| ALDH2 | rs671 | g -> a | ALDH 2*2: E -> K | 12: 112241766 | Sensibilité à l'alcool: Allele ALDH2*2 (a) is associated with a very high incidence of acute alcohol intoxication in Orientals and South American Indians, as compared to Caucasians. Seong-Jin Kim est hétérozygote G/A: il ne doit pas trop bien supporter l'alcool.... |
UniProtKB P05091 | Alfred |
| MCM6 | rs4988235 | c -> t (gène lu 'à l'envers') | non codant; influence l'expression de la lactase (LCT) | 2: 136608646 | Intolérance au lactose (c/c) ; C. Venter n'est pas intolérant au lactose, par contre J. Watson l'est à moitié... | UniProtKB P09848 | Alfred |
* Les positions en bases sur les chromosomes peuvent changer avec les nouvelles versions du génome (Releases ou Genome Build) de référence. L'entrée dbSNP permet de retrouver l'info actualisée. En janvier 2012 c'est 37.3 qui est actuel.
*2 Alicia Sanchez Mazas précise : On n’est pas noir ou blanc, on est plus ou moins foncé. Il s’agit d’un caractère quantitatif, à variation continue. La mutation dans SLC24A5 est l’une des mutations qui interviennent dans la régulation de la production de mélanine et la variation, quantitative, de ce caractère.
SNP populaires selon SNPedia
Ce document fait partie des formations continues BIST.
 1E) Exemple d'exercice de type "enquête".
1E) Exemple d'exercice de type "enquête".
Exemple d'exercice de type "enquête" : des séquences ADN sont proposées et il faut retrouver les patients dont les phénotypes sont donnés. ici.pdf
Un site sur DNA ancestry (Généalogie génétique)
Une publication Mitochondrial DNA analysis of the putative heart of Louis XVII, son of Louis XVI and Marie-Antoinette. Vous pouvez aller voir les positions testées pour prouver la filiation sur la séquence de la mitochondrie (appelée chrM) à UCSC GeneTests
Comparer (en terme de nombre de polymorphismes connus) une région génomique de même longueur 200-300 pb avec des indices de diversités moléculaires très différents. Le nombre de polymorphismes SNP est visible grâce au genome browser UCSC ce qui permet d'estimer visuellement la diversité.
1- Région codante avec une diversité moléculaire (
sélection stabilisante
, balancée); beaucoup de polymorphismes: maintient la diversité
Exemple: HLA-A (exons 2 et 3) impliqués dans la réponse immune:
exon 2: coller dans
UCSC
chr6:29,910,998-29,911,443 ;
exon 3: coller dans
UCSC
chr6:29,911,045-29,911,118
2- Région codante sous sélection adaptative (en fonction du milieu: une mutation peut être délétère). Ces gènes contiennent peu de polymorphisme:
Exemple: histone H4: coller dans UCSC chr1:149,804,221-149,804,615
3- Région non codante: accumulation de
mutations au hasard
(souvent utilisé pour reconstruire l'histoire d'une population)
Exemple: la région HVS1 (Hypervariable segment 1) de l'ADN de mitochondrie:coller dans UCSC chrM:16,024-16,365
NB: tous les polymorphismes connus ne sont pas répertoriés dans dbSNP (voir question suivante)
Voir aussi la diversité génétique des élèves genevois sur cette séquence HVS1 justement :
Séquençage d’élèves dans Expériment@l !

 Contexte biologique sur les HLA et la diversité génétique:
Contexte biologique sur les HLA et la diversité génétique:
Banque de données spécialisées (IMGT/HLA Database) pour les séquences HLA humaines Elle répertorie tous les allèles connus (combinaison de polymorphismes) chez l'homme (mais pas leur fréquence...)
Select the feature to align : - HLA-A exon 2: région de l'exon 2 particulièrement polymorphique (impliquée dans la réponse immune)
On peut retrouver cette région @ UCSC aux positions: chr6:29,910,534-29,910,607
On observe qu'il y a un très grand nombre de polymorphismes connus
- HLA-A exon 3: région de l'exon 3 particulièrement polymorphique (impliquée dans la réponse immune)
On peut retrouver cette région @ UCSC aux positions: chr6:29,911,045-29,911,118
- HLA-DRA: région peu polymorphique :
On peut retrouver cette région @ UCSC aux positions: chr6:32,410,225-32,410,470
On observe qu'il y a très peu de polymorphismes connus
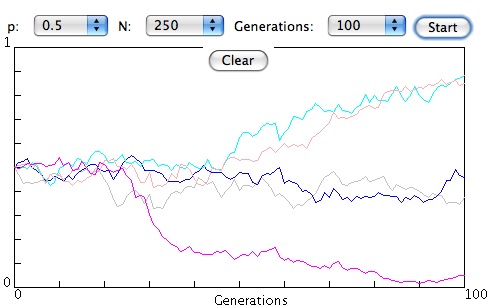
En partant d'une fréquence de 0.5, on peut simuler plusieurs fois l'évolution de la fréquence en fonction des générations et observer la dérive génétique.
On peut varier la taille de la population pour voir que la dérive est plus marquée avec une petite population.

les cartes sont ici : HLA allele frequency maps: A , C , B , DRB1 , DQA1 and DQB1 , DPA1 and DPB1 .
Certains de ces allèles sont associés à des maladies,
Pour explorer au niveau des populations, GenAlex est ce qu'il y a de plus user-friendly . Avec cette macro pour Excel, on peut faire plusieurs exercices qui travaillent soit sur des fréquences d'allèles, soit sur des séquences, des choses très simples, mais assez parlantes.
Alicia Sanchez-Mazas propose un exercice simple d’analyse de données génétiques :
Autres liens utiles
Retour à BIST | Swiss-Prot | M.C. Blatter | Projets Home de F. Lo
{kind=link}
{kind=link}